
BTX-A51, a novel, small molecule multi-specific inhibitor of CK1α, CDK7 and CDK9, promotes cancer cell apoptosis through a synergistic mechanism of action that targets multiple master regulators of cancer.
p53 is a master regulator of programmed cell death (apoptosis) and the DNA damage response. Suppressing the function of p53 is a common mechanism of cancer cell survival. Casein kinase 1α (CK1α), and the cyclin-dependent kinases 7 and 9 (CDK7 and CDK9) work synergistically in tumor cells to inhibit p53 via downregulation of mouse double minute 2 homolog (MDM2), a protein which mediates the degradation of p53.1 Preclinical data in acute myeloid leukemia (AML), liposarcoma and breast cancer cell lines demonstrate that BTX-A51 robustly increases p53 levels while decreasing expression of MDM2 and myeloid cell leukemia-1 (MCL-1), resulting in programmed cell death.1

BTX-A51 inhibits the expression of MDM2 and MCL-1 and increases the expression of p53, resulting in apoptosis.
BTX-A51 Leverages Cancer Cells’ Addiction to Super Enhancers
Super Enhancers (SEs) are master regulators of the transcription of key oncogenes whose proteins are involved in pathways essential for the proliferation and survival of cancer cells, including MDM2 and MCL-1. SEs rely on CDK7 and CDK9 to activate RNA polymerase II to over-express these oncogenes—and are therefore highly sensitive to CDK7/9 inhibition. Preclinical data in AML, breast and liposarcoma demonstrate that BTX-A51 preferentially decreases expression of the oncogenes necessary for cancer cell survival, resulting in selective apoptosis of cancer cells.1,2
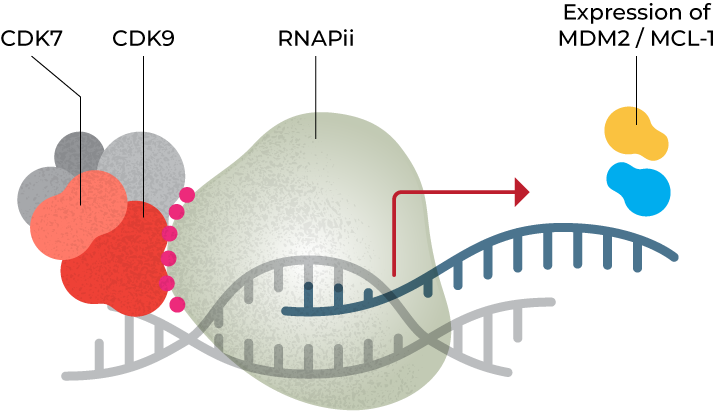

CDK7 & CDK9 phophorylate RNA polymerase II at Super Enhancers to stimulate transcription; inhibiting CDK7 & CDK9 reduces expression of products such as MDM2 and MCL-1
Ablation of MDM2 Leads to Accumulation of p53
MDM2 is an E3 ubiquitin ligase whose principal function is to regulate the turnover of p53. MDM2 adds ubiquitin to p53 marking it for degradation by the proteasome. Abrogation of MDM2 expression by blocking SE-mediated transcription leads to accumulation of p53. Direct MDM2 inhibitors have been investigated clinically and shown to have anticancer activity in tumors that overexpress MDM2, but as a class direct MDM2 inhibitors cause dose-limiting thrombocytopenia and neutropenia that limit their utility. BTX-A51 inhibits MDM2 indirectly, and this mechanism has not been associated with the same adverse event profile as direct inhibitors.2 Early clinical data suggests that BTX-A51 increases p53 and activates apoptosis while maintaining a favorable safety profile.2
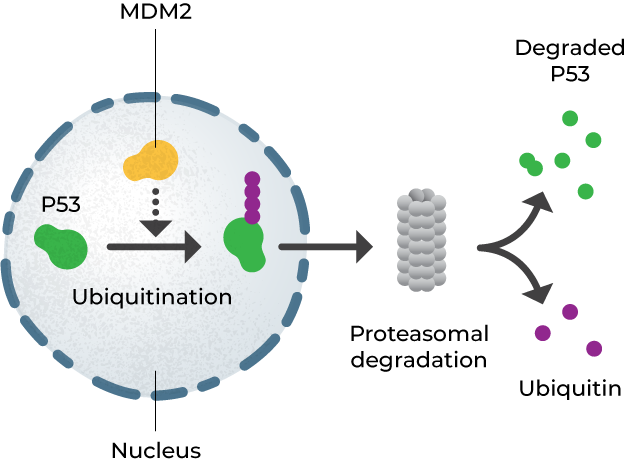

MDM2 increases the degradation of p53 via ubiquitination; blocking MDM2 expression leads to p53 accumulation
Ablation of MCL-1 Blocks a Major Mechanism for Resistance in AML and Solid Tumors
MCL-1 enhances tumor cell survival by binding peptides that activate the first steps of apoptosis. In AML, MCL-1 overexpression is a major mechanism of resistance to venetoclax-based therapeutic regimens.4 MCL-1 has a short half-life and must be continually synthesized by the tumor cell, starting with SE-mediated transcription. BTX-A51 blocks this expression, leading to loss of MCL-1. Direct MCL-1 inhibitors have been tested clinically and shown to have anti-cancer activity, but they have also demonstrated dose limiting toxicity, including cardiotoxicity in patients. Since BTX-A51 inhibits MCL-1 at the transcriptional level, it acts differently than direct MCL-1 inhibitors. Phase 1 studies with BTX-A51 in both AML and solid tumors patients have demonstrated no evidence of cardiotoxicity at pharmacologically active dose levels.3


MCL-1 sequesters peptides that would otherwise stimulate apoptosis; blocking MCL-1 expression enables a key step in apoptosis, the release of cytochrome C from mitochondria
CDK7 is a Master Regulator of Tumor Cell Growth
CDK7 is a key regulator of downstream cyclin-dependent kinases that control completion of cell cycle steps necessary for cell growth. This includes CDK4, CDK6, CDK2 and CDK1, which are all activated through phosphorylation by CDK7. CDK7 also regulates the phosphorylation of Rb, a transcription repressor, to enable the expression of genes essential for tumor cell growth. Thus, in addition to the effect on Super Enhancer-mediated gene expression, inhibition of CDK7 blocks tumor cell growth by inhibiting cell cycle progression.5


CDK7 regulates downstream cyclin depenent kinases through its CAK activity; inhibiting CDK7 interferes with the cell cycle and inhibits tumor cell growth
References
Minzel W, Venkatachalam A, Fink A, Hung E, Brachya G, Burstain I, et al. Small Molecules Co-targeting CKIα and the Transcriptional Kinases CDK7/9 Control AML in Preclinical Models. Cell. 2018;175(1):171–185.
Liu R., Solimini NL, Bhola P, Branigan TB, et al. Targeting casein kinase 1 alpha (CK1 alpha) and transcriptional CDKs (CDK7/9) in human liposarcomas Cancer Res 2024; 84 (6Suppl): Abstract nr 604.
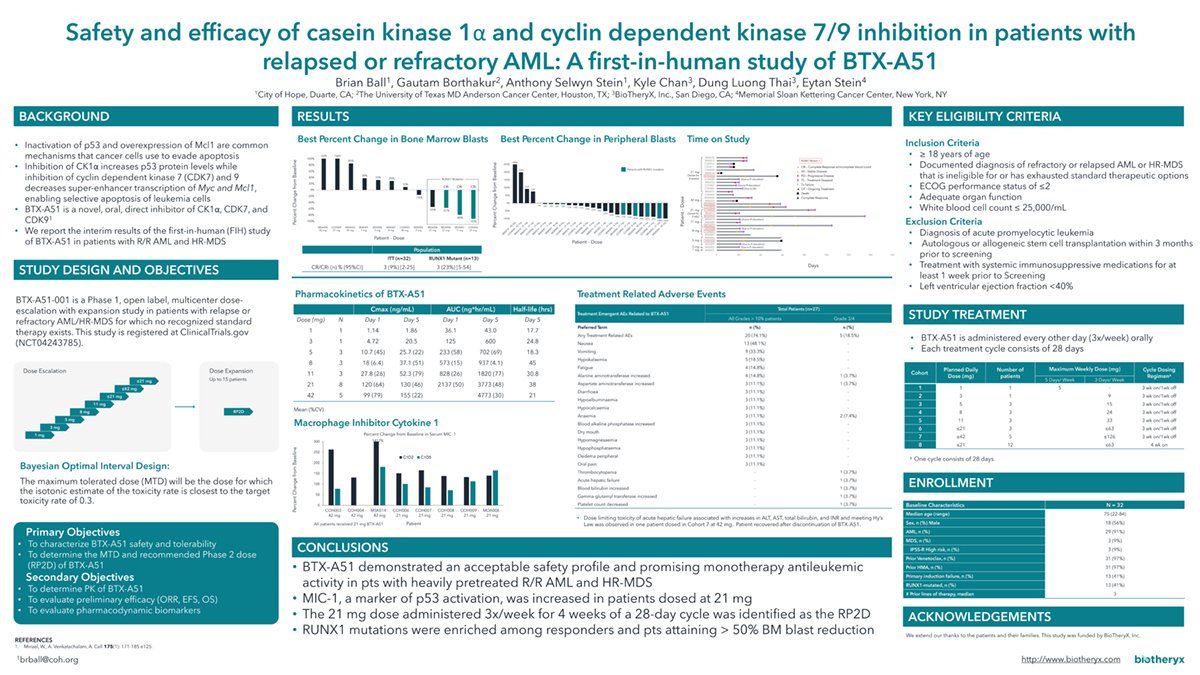
Ball B, Borthakur G, Stein AS, Chan K, Thai DL, and Stein E. Safety and efficacy of casein kinase 1α and cyclin dependent kinase 7/9 inhibition in patients with relapsed or refractory AML: A first-in-human study of BTX-A51. J Clin Oncol 40, 2022 (suppl 16; abstr 7030).
Liu VM, Guièze R, Rosebrock D, Jourdain AA, Hernández-Sánchez M, Martinez AZ, et al.. MCL-1 and PKA/AMPK axis fuel venetoclax resistance in lymphoid cancers. Blood (2019) 134:1284–4.
Coombes RC, Howell, S, Lord, SR, Kenny L, Mansi, J, Mitri Z, et al. Dose escalation and expansion cohorts in patients with advanced breast cancer in a Phase I study of the CDK7-inhibitor samuraciclib. Nat Commun 14, 4444 (2023).
Poster Presentations

Targeting casein kinase 1 alpha (CK1 alpha) and transcriptional CDKs (CDK7/9) in human liposarcomas.
Presented at: 2024 AACR Annual Meeting
Safety and efficacy of casein kinase 1α and cyclin dependent kinase 7/9 inhibition in patients with relapsed or refractory AML: A first-in-human study of BTX-A51.
Presented at: 2022 ASCO Annual Meeting